“戈谢病”在大众里一定很陌生,甚至一些医生都不知道。它是罕见病中的罕见病,全国确诊的只有300多人。患者内脏肿大,直接表现就是大肚子,会导致疼痛、骨损伤甚至死亡。江苏目前确诊的也就四五例,其中徐州3岁女童熙熙(化名)就是其中一名。
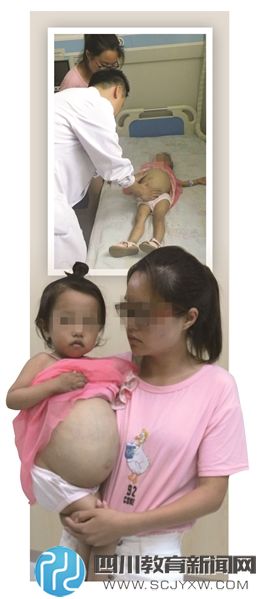
有记者来到徐州市儿童医院,采访了正在接受治疗的熙熙,孩子肚子已经大得像皮球,甚至随时可能胀破,生命岌岌可危。而尽管治疗戈谢病的药物已在我国药监局注册并上市,但费用昂贵,一年药费达60万元以上,且需终身用药。江苏血液病专家呼吁,如戈谢病这样的“罕见病”,一些省市已纳入医保,希望我省也能将这样的罕见病药物纳入大病医保。
求医一波三折
最终在北京确诊罕见病
王成和张金金都是江苏沛县河口镇人,2013年4月12日迎来了宝贝女儿小熙熙。80后的王成是一家网店的运营主管,90后的妻子张金金在家照顾女儿,原本这个三口之家拥有很多幸福的可能,但一切皆因女儿突如其来的怪病戛然而止。
记者了解到,和大多数戈谢病患者一样,小熙熙的确诊之路也是一波三折。2014年除夕,熙熙发烧到40度,夫妻俩连夜将她送往县医院。经过血常规检查发现,熙熙的血小板还不到正常值的一半。
与此同时,张金金发现女儿的肚子越来越大,且右部明显发硬。本来他们也没太在意,因为多数婴儿的小肚子都是挺挺的。不过同村一位在沛县县医院工作的医生给熙熙摸诊后,觉得并不乐观,建议他们去医院做B超。
于是,一家人带着熙熙来到徐州市儿童医院。医生起先怀疑熙熙是白血病,谁知住了八九天院后,仍无法检查出病因,医生建议他们到苏州大学附属儿童医院检查,因为那里治疗血液病的水平全国领先。于是,2014年5月,夫妻二人带着熙熙前往苏州,做了骨髓穿刺等多项检查。
来到苏州的第二天,熙熙便检查出了戈谢细胞的存在。不仅家长来说,“戈谢病”这个名称很陌生,甚至连当时在苏州儿童医院实习的医学院学生也第一次听说。
王成至今还记得,血液科的主任告诉他们检测出戈谢细胞时,脸上流泪出那种无奈的表情,“医生说,宝宝体内有一种酶无法代谢,脾脏和肝脏会越来越大。这种病一旦确诊了,一般情况下是没得治了,除非每年花上近百万的医药费。”抱着最后一丝侥幸,夫妻俩带着孩子北上检测。结果熙熙于2014年7月8日这天在北京协和医院最终确诊为戈谢病。
之后,一家人带着孩子在北京的一家医院想做骨髓移植试试看,但是就在住院期间,孩子查出来肺部有真菌感染,经过几个月治疗都没好转,无法进行移植。加上四处奔波,孩子的治疗费用基本上已经用完,一家人只能带着小熙熙回到徐州。
病情慢慢恶化
孩子肚皮胀大快要撑破了
从北京回来后,熙熙回家进行一些维持的治疗,而肚子是一天比一天大,熙熙别说出门了,她连蹲都蹲不下来,正常的大小便都无法完成。这段时间,熙熙的病情加重了,王成告诉记者,孩子总是抱着肚子喊疼。“她每天晚上都会大哭,我在网上看到,有大一些得病的孩子会形容,那种疼痛感像刀割一样,熙熙不会表达,但她的痛苦可想而知。”由于担心孩子是脾脏梗阻了,7月22日中午,当地气温已经达到35度的高温,夫妻俩还是不放心,带着孩子,坐了一个小时的车来到徐州市儿童医院。
记者当天也赶到徐州市儿童医院,等到夫妻俩赶到医院的时候,已经是接近下午1点,一直等在门诊的普外科魏建民主任接诊了小熙熙。
熙熙比父母向记者求助时提供的照片更加严重,孩子的身材比同龄孩子要矮上一大截,腹部已经像是足月快临盆孕妇,肚皮布满血筋,仿佛很快就要被撑破了!记者用手触摸,仿佛石头般硬,但熙熙的胳膊和腿都非常细,只有同龄孩子的一半。医生为入院的小熙熙称了下体重,只有11公斤,严重的营养不良。而医生告诉记者,这11公斤里面,那个大得快撑破肚皮的脾脏估计就有5公斤!
“孩子现在不能吃任何东西,只能喝一些配方奶粉,一次也就60毫升左右,喝一点点就觉得肚子胀得难受。”说到这里,孩子的妈妈张金金哭了。
在简单的检查之后,普外科魏建民主任医师告诉记者,熙熙的脾脏已经肿大到将盆腔占满了;肿大的肝脏被挤到一边,质地比较硬,怀疑有轻度的肝硬化。孩子之所以这么瘦小,是因为疾病影响到骨骼发育,如果再不治疗,可能今后个头也不会长了。
魏主任建议先将熙熙收治住院,还要完善进一步检查,并且请血液科等专家进行会诊。记者发现,整个全过程中,小熙熙都非常乖巧,配合医生的检查,还会笑着安慰妈妈说,“妈妈我现在不疼了,我不怕。”张金金说,孩子对这里已经非常熟悉了,孩子总是感冒发烧,几乎每一两个月,熙熙就要到徐州市儿童医院住一次院,手上胳膊上都是针眼,但是每次她都默默忍受。
治疗费用非常昂贵
眼下每年要花60万
孩子长大后治疗费超百万
戈谢病到底是一种什么样的疾病?血液病专家告诉记者,它是一种最常见的溶酶体贮积症,属于常染色体隐性方式遗传,因位于1 号染色体长臂上的葡萄糖脑苷脂酶基因的突变,造成体内该酶的缺乏,细胞内脂类分子无法正常代谢而堆积在溶酶体中。导致各种临床表现如肝脾肿大、贫血、血小板减少以及骨骼病变等各种临床表现。
戈谢病是罕见病中的罕见病,戈谢病的发病率目前在国内没有明确数据,大约是二十万分之一。目前我国内地确诊的戈谢病患者大概在370例左右,而江苏目前报道的戈谢病患者只有四五例。
“我们国家真正明确诊断的戈谢病,远远比国际公布的发病率要低,原因之一的就是,有的患儿因为症状不是很重,最初症状可能就是贫血、脾大什么的,有的孩子可能就漏诊了;还有一种就是,症状特别重,还没等诊断清楚,患儿可能就已经去世了。”昨天,血液病专家、苏州大学附属儿童医院血液科主任胡绍燕教授告诉记者,所以,临床她们见到戈谢病病人很少。
虽然戈谢病罕见难治,可也并非无药可治。胡绍燕教授告诉记者,酶替代疗法是目前国内治疗戈谢病的金标准,具体做法是通过两周一次的常规静脉滴注来替代缺少的酶。
据介绍,注射用的伊米苷酶即“思而赞”,是我国目前唯一能有效治疗戈谢病的药物,但这种药物价格不菲。而且,戈谢病患者的用药剂量需要根据患者的体重以及疾病的严重程度。按照正常剂量,现在熙熙每月需要注射2支药,这样算下来,差不多每年需要60万元的治疗费用,并且随着年龄的增长,孩子用药量要增加,每年的治疗费用或超过百万。这对王成一家来说,简直无法想象。
专家呼吁
将戈谢病纳入医保
“我们知道,这个病不是没法儿治,听医生说,如果用上药物,脾脏会慢慢变小,孩子慢慢会和其他正常小孩儿一样,可是费用太高了,我们没办法。”熙熙的父亲王成告诉记者,他们曾经向媒体求助过,也受到了一些社会捐助,他们一家非常感激帮助他们的好心人,但是孩子的病需要一生都维持治疗,所以目前他们更希望是这种疾病的药物可以纳入医保和民政救助范围内。
王成从戈谢病病友那里得知,青岛等地已经出台相关政策救助戈谢病患者,但江苏并未对这一罕见病出台相关政策。受此启发,他开始了自救之路。“这两年我们南京前前后后不知道跑了多少趟,医保、卫生、民政都去过,”王成说,他们得到回复都是需要审批。
据悉,上海、昆明、宁夏、青岛等地,已经出台了针对戈谢病患者的医疗保障政策。2016年1月1日起,浙江也将戈谢病纳入了医保。苏州大学附属儿童医院血液科主任胡绍燕教授也呼吁说,希望政府尽早将戈谢病纳入医保范畴,从制度和政策上保证戈谢病群体的长期治疗。要知道对于戈谢病人来说,只要终身用药,生活就和正常人无异,而这对于戈谢病家庭来说,却是弥足珍贵的。







